


Технологию редактирования генома планируют использовать для лечения детей с синдромом раннего старения
Прогерия – очень редкое генетическое заболевание с симптомами раннего старения: дети с таким диагнозом обычно живут не больше 14 лет, чаще всего умирая от сердечного приступа или инсульта. Результаты генной терапии лабораторных животных с прогерией вселяют надежду на возможность лечения детей с этой врожденной болезнью, причем уже в ближайшем будущем
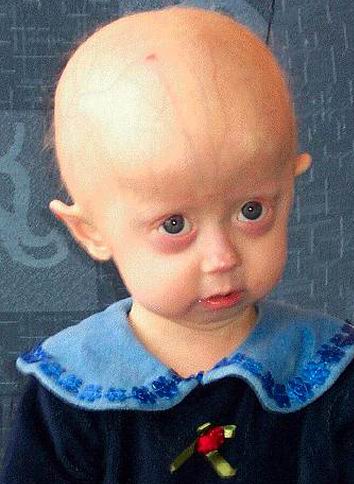
Детской формой прогерии – синдромом Хатчинсона-Гилфорда – в мире страдает около 400 человек. У детей с этим редчайшим заболеванием уже с раннего возраста наблюдаются задержка роста, черепно-лицевая диспропорция (большой череп и маленькое лицо), морщинистая жесткая кожа, облысение, изменения суставов, остеопороз и атеросклероз, т.е. все признаки старения.
Одна из основных причин этой болезни – мутация в гене LMNA, при которой в ДНК пара азотистых оснований гуанин–цитозин заменяется на аденин–тимин. Этот ген кодирует белок ламин А, обеспечивающий правильное формирование мембраны клеточного ядра и генетического материала клетки. Дефектный белок вызывает нарушения пространственной организации ядерной ДНК, что при клеточном делении приводит к многочисленным ошибкам.
 Уже были предприняты попытки «исправить» мутантный ген у лабораторных мышей с прогерией при помощи широко используемой технологии редактирования генома CRISPR. Однако результаты оказались неудовлетворительными: здоровье животных улучшалось незначительно, а риск побочных явлений в виде нецелевого редактирования генов был велик.
Уже были предприняты попытки «исправить» мутантный ген у лабораторных мышей с прогерией при помощи широко используемой технологии редактирования генома CRISPR. Однако результаты оказались неудовлетворительными: здоровье животных улучшалось незначительно, а риск побочных явлений в виде нецелевого редактирования генов был велик.
Недавно исследователи из Гарвардского университета (США) использовали для этой цели немного другой инструментарий – так называемый редактор адениновых оснований. В основе этой технологии лежит модифицированная система CRISPR, к которой пришиты ферменты – адениновые деаминазы. CRISPR находит на ДНК нужный участок, а затем с помощью ферментов «неправильный» аденин преобразуется в «исходный» гуанин. При этом, в отличие от обычного редактирования, разрезаются не обе нити двуцепочечной ДНК, а лишь одна.
Эту систему редактирования генома «упаковали» в безопасный аденоассоциированный вирус (он инфицирует клетки человека и некоторых других приматов, но заболевания не вызывает) – стандартное транспортное средство генной терапии, и ввели двухнедельным мышам с прогерией.
Результаты превзошли ожидания: через полгода после инъекции генно-инженерных вирусов уровень мутантного белка в скелетных мышцах, сердце, аорте и печени упал, а нормального ламина А – повысился. В аорте подопытных мышей практически отсутствовали признаки роста фиброзной ткани и потери гладкомышечных клеток, характерные для этой болезни. Животные не страдали от сердечно-сосудистой патологии и прожили вдвое дольше своих собратьев с такой же мутацией.
Есть, правда, одно «но»: у некоторых особей в печени появились опухоли. Такое и ранее наблюдалось у мышей, получавших генную терапию с высокими дозами аденоассоциированного вируса. У людей такие последствия не описаны, но ученые сейчас работают над тем, чтобы снизить количество вводимых вирусных частиц.
Одновременно предпринимаются шаги по применению имеющейся версии генной терапии на больных детях. Предварительные исследования на культурах клеток соединительной ткани – фибробластах, полученных от больных, показывают, что такая коррекция патологического генного варианта как минимум достаточно эффективна, а признаки нецелевого редактирования генома отсутствуют.
Возможно, дети с прогерией получат новую терапию уже в недалеком будущем: каким бы ни поспешным казался такой шаг, терять им практически нечего.
Фото: https://commons.wikimedia.org, https://commons.wikimedia.org








